[med-svn] [Git][med-team/unanimity][master] 4 commits: Add myself as Uploader
Andreas Tille
gitlab at salsa.debian.org
Thu Oct 11 12:59:37 BST 2018
Andreas Tille pushed to branch master at Debian Med / unanimity
Commits:
c6810af3 by Andreas Tille at 2018-10-11T07:14:05Z
Add myself as Uploader
- - - - -
894b77df by Andreas Tille at 2018-10-11T07:34:39Z
Remove references to non-existing images
- - - - -
5d696741 by Andreas Tille at 2018-10-11T07:35:49Z
hardening=+all
- - - - -
2eaa1f1a by Andreas Tille at 2018-10-11T08:47:56Z
Add manpages (except for arrow which does not have any sensible output)
- - - - -
8 changed files:
- + debian/ccs.1
- debian/control
- + debian/createmanpages
- + debian/gcpp.1
- + debian/manpages
- + debian/patches/do_not_refer_to_non-existing_images.patch
- debian/patches/series
- debian/rules
Changes:
=====================================
debian/ccs.1
=====================================
@@ -0,0 +1,97 @@
+.\" DO NOT MODIFY THIS FILE! It was generated by help2man 1.47.7.
+.TH CCS "1" "October 2018" "ccs 3.1.0" "User Commands"
+.SH NAME
+ccs \- Generate circular consensus sequences from subreads
+.SH SYNOPSIS
+.B ccs
+[\fI\,options\/\fR] \fI\,INPUT OUTPUT\/\fR
+.SH DESCRIPTION
+Generate circular consensus sequences (ccs) from subreads.
+.SH OPTIONS
+.TP
+\fB\-h\fR,\-\-help
+Output this help.
+.TP
+\fB\-\-log\-level\fR,\-\-logLevel
+Set log level. ["INFO"]
+.TP
+\fB\-\-version\fR
+Output version info.
+.TP
+\fB\-\-force\fR
+Overwrite OUTPUT file if present.
+.TP
+\fB\-\-zmws\fR
+Generate CCS for the provided comma\-separated holenumber ranges only. Default = all
+.TP
+\fB\-\-maxLength\fR
+Maximum length of subreads to use for generating CCS. [21000]
+.TP
+\fB\-\-minLength\fR
+Minimum length of subreads to use for generating CCS. [10]
+.TP
+\fB\-\-minPasses\fR
+Minimum number of subreads required to generate CCS. [3]
+.TP
+\fB\-\-minPredictedAccuracy\fR
+Minimum predicted accuracy in [0, 1]. [0.9]
+.TP
+\fB\-\-minIdentity\fR
+Minimum identity to the POA to use a subread. 0 disables this filter. [0.82]
+.TP
+\fB\-\-minZScore\fR
+Minimum z\-score to use a subread. NaN disables this filter. [\-3.4]
+.TP
+\fB\-\-maxDropFraction\fR
+Maximum fraction of subreads that can be dropped before giving up. [0.34]
+.TP
+\fB\-\-minSnr\fR
+Minimum SNR of input subreads. [3.75]
+.TP
+\fB\-\-minReadScore\fR
+Minimum read score of input subreads. [0.75]
+.TP
+\fB\-\-byStrand\fR
+Generate a consensus for each strand.
+.TP
+\fB\-\-noPolish\fR
+Only output the initial template derived from the POA (faster, less accurate).
+.TP
+\fB\-\-polish\fR
+Emit high\-accuracy CCS sequences polished using the Arrow algorithm
+.TP
+\fB\-\-polishRepeats\fR
+Polish repeats of 2 to N bases of 3 or more elements. [0]
+.TP
+\fB\-\-richQVs\fR
+Emit dq, iq, and sq "rich" quality tracks.
+.TP
+\fB\-\-reportFile\fR
+Where to write the results report. ["ccs_report.txt"]
+.TP
+\fB\-\-modelPath\fR
+Path to a model file or directory containing model files.
+.TP
+\fB\-\-modelSpec\fR
+Name of chemistry or model to use, overriding default selection.
+.TP
+\fB\-\-numThreads\fR
+Number of threads to use, 0 means autodetection. [0]
+.TP
+\fB\-\-logFile\fR
+Log to a file, instead of STDERR.
+.TP
+\fB\-\-emit\-tool\-contract\fR
+Emit tool contract.
+.TP
+\fB\-\-resolved\-tool\-contract\fR
+Use args from resolved tool contract.
+.SS "Arguments:"
+.TP
+input
+Input file.
+.TP
+output
+Output file.
+.SH AUTHOR
+This manpage was written by Andreas Tille for the Debian distribution and can be used for any other usage of the program.
=====================================
debian/control
=====================================
@@ -1,6 +1,7 @@
Source: unanimity
Maintainer: Debian Med Packaging Team <debian-med-packaging at lists.alioth.debian.org>
Uploaders: Afif Elghraoui <afif at debian.org>
+ Andreas Tille <tille at debian.org>
Section: science
Testsuite: autopkgtest-pkg-python
Priority: optional
@@ -41,7 +42,6 @@ Description: generate and process accurate consensus nucleotide sequences
Package: python-consensuscore2
Architecture: any
Section: python
-Testsuite: autopkgtest-pkg-python
Depends: ${shlibs:Depends},
${misc:Depends},
${python:Depends}
=====================================
debian/createmanpages
=====================================
@@ -0,0 +1,33 @@
+#!/bin/sh
+MANDIR=debian
+mkdir -p $MANDIR
+
+VERSION=`dpkg-parsechangelog | awk '/^Version:/ {print $2}' | sed -e 's/^[0-9]*://' -e 's/-.*//' -e 's/[+~]dfsg$//'`
+NAME=`grep "^Description:" debian/control | sed 's/^Description: *//' | head -n1`
+PROGNAME=`grep "^Package:" debian/control | sed 's/^Package: *//'`
+
+AUTHOR=".SH AUTHOR\nThis manpage was written by $DEBFULLNAME for the Debian distribution and
+can be used for any other usage of the program.
+"
+
+# If program name is different from package name or title should be
+# different from package short description change this here
+progname=ccs
+help2man --no-info --no-discard-stderr \
+ --name="Generate circular consensus sequences from subreads" \
+ --version-string="$VERSION" ${progname} > $MANDIR/${progname}.1
+echo $AUTHOR >> $MANDIR/${progname}.1
+
+progname=gcpp
+help2man --no-info --no-discard-stderr \
+ --name="Compute genomic consensus from alignments and call variants relative to the reference" \
+ --version-string="$VERSION" ${progname} > $MANDIR/${progname}.1
+echo $AUTHOR >> $MANDIR/${progname}.1
+
+echo "$MANDIR/*.1" > debian/manpages
+
+cat <<EOT
+Please enhance the help2man output.
+The following web page might be helpful in doing so:
+ http://liw.fi/manpages/
+EOT
=====================================
debian/gcpp.1
=====================================
@@ -0,0 +1,241 @@
+.\" DO NOT MODIFY THIS FILE! It was generated by help2man 1.47.7.
+.TH GCPP "1" "October 2018" "gcpp 3.1.0" "User Commands"
+.SH NAME
+gcpp \- Compute genomic consensus from alignments and call variants relative to the reference
+.SH SYNOPSIS
+.B gcpp
+[\fI\,options\/\fR] \fI\,INPUT\/\fR
+.SH DESCRIPTION
+Compute genomic consensus from alignments and call variants relative to the reference.
+.SS "Basic required options:"
+.TP
+\fB\-\-referenceFilename\fR,\-\-reference,\-r
+The filename of the reference FASTA
+file.
+.TP
+\fB\-\-outputFilenames\fR,\-o
+The output filename(s), as a
+comma\-separated list. Valid output
+formats are .fa/.fasta, .fq/.fastq,
+\&.gff, .vcf
+.SS "Parallelism:"
+.TP
+\fB\-\-numThreads\fR,\-j
+The number of threads to be used.
+[1]
+.SS "Output filtering:"
+.TP
+\fB\-\-minConfidence\fR,\-q
+The minimum confidence for a variant
+call to be output to
+variants.{gff,vcf} [40]
+.TP
+\fB\-\-minCoverage\fR,\-x
+The minimum site coverage that must
+be achieved for variant calls and
+consensus to be calculated for a
+site. [5]
+.TP
+\fB\-\-noEvidenceConsensusCall\fR
+The consensus base that will be
+output for sites with no effective
+coverage. ["lowercasereference"]
+.SS "Read selection/filtering:"
+.TP
+\fB\-\-coverage\fR,\-X
+A designation of the maximum
+coverage level to be used for
+analysis. Exact interpretation is
+algorithm\-specific. [100]
+.TP
+\fB\-\-minAccuracy\fR
+The minimum acceptable window\-global
+alignment accuracy for reads that
+will be used for the analysis
+(arrow\-only). [0.82]
+.TP
+\fB\-\-minMapQV\fR,\-m
+The minimum MapQV for reads that
+will be used for analysis. [10]
+.TP
+\fB\-\-minReadScore\fR
+The minimum ReadScore for reads that
+will be used for analysis
+(arrow\-only). [0.65]
+.TP
+\fB\-\-minSnr\fR
+The minimum acceptable
+signal\-to\-noise over all channels for
+reads that will be used for analysis
+(arrow\-only). [3.75]
+.TP
+\fB\-\-minZScore\fR
+The minimum acceptable z\-score for
+reads that will be used for analysis
+(arrow\-only). [\-3.4]
+.TP
+\fB\-\-barcode\fR,\-\-barcodes
+Comma\-separated list of barcode
+pairs to analyze, either by name,
+such as 'lbc1\-\-lbc1', or by index,
+such as '0\-\-0'. NOTE: Filtering
+barcodes by name requires a barcode
+file.
+.TP
+\fB\-\-barcodeFile\fR
+Fasta file of the barcode sequences
+used. NOTE: Only used to find barcode
+names
+.TP
+\fB\-\-referenceWindow\fR,\-\-referenceWindows,\-w
+The window (or multiple
+comma\-delimited windows) of the
+reference to be processed, in the
+format refGroup:refStart\-refEnd
+(default: entire reference).
+.TP
+\fB\-\-referenceWindowsFile\fR,\-W
+A file containing reference window
+designations, one per line
+.SS "Algorithm and parameter settings:"
+.TP
+\fB\-\-algorithm\fR
+The consensus algorithm used.
+["arrow"]
+.TP
+\fB\-\-maskRadius\fR
+Radius of window to use when
+excluding local regions for exceeding
+maskMinErrorRate, where 0 disables
+any filtering (arrow\-only). [0]
+.TP
+\fB\-\-maskErrorRate\fR
+Maximum local error rate before the
+local region defined bymaskRadius is
+excluded from polishing (arrow\-only).
+[0]
+.TP
+\fB\-\-parametersFile\fR,\-P
+Parameter set filename (such as
+ArrowParameters.json or
+QuiverParameters.ini), or directory D
+such that either
+D/*/GenomicConsensus/QuiverParameters
+\&.ini, or
+D/GenomicConsensus/QuiverParameters.i
+ni, is found. In the former case,
+the lexically largest path is chosen.
+.TP
+\fB\-\-parametersSpec\fR,\-p
+Name of parameter set
+(chemistry.model) to select from the
+parameters file, or just the name of
+the chemistry, in which case the best
+available model is chosen. Default
+is 'auto', which selects the best
+parameter set from the alignment data
+["auto"]
+.TP
+\fB\-\-maxIterations\fR
+Maximum number of iterations to
+polish the template. [40]
+.TP
+\fB\-\-maxPoaCoverage\fR
+Maximum number of sequences to use
+for consensus calling. [11]
+.TP
+\fB\-\-mutationSeparation\fR
+Find the best mutations within a
+separation window for iterative
+polishing. [10]
+.TP
+\fB\-\-mutationNeighborhood\fR
+Find nearby mutations within
+neighborhood for iterative polishing.
+[10]
+.TP
+\fB\-\-readStumpinessThreshold\fR
+Filter out reads whose aligned
+length along a subread is lower than
+a percentage of its corresponding
+reference length. [0.1]
+.SS "Verbosity and debugging:"
+.TP
+\fB\-\-logFile\fR
+Log to a file, instead of STDERR.
+.TP
+\fB\-\-dumpEvidence\fR,\-d
+Dump evidence data
+.TP
+\fB\-\-evidenceDirectory\fR
+Directory to dump evidence into.
+.TP
+\fB\-\-annotateGFF\fR
+Augment GFF variant records with
+additional information
+.TP
+\fB\-\-reportEffectiveCoverage\fR
+Additionally record the
+*post\-filtering* coverage at variant
+sites
+.SS "Advanced configuration options:"
+.TP
+\fB\-\-referenceChunkSize\fR,\-C
+Size of reference chunks. [500]
+.TP
+\fB\-\-referenceChunkOverlap\fR
+Size of reference chunk overlaps.
+[5]
+.TP
+\fB\-\-simpleChunking\fR
+Disable adaptive reference chunking.
+.TP
+\fB\-\-diploid\fR
+Enable detection of heterozygous
+variants (experimental)
+.TP
+\fB\-\-fast\fR
+Cut some corners to run faster.
+Unsupported!
+.TP
+\fB\-\-skipUnrecognizedContigs\fR
+Do not abort when told to process a
+reference window (via
+\fB\-w\fR/\-\-referenceWindow[s]) that has no
+aligned coverage. Outputs emptyish
+files if there are no remaining
+non\-degenerate windows. Only
+intended for use by smrtpipe
+scatter/gather.
+.TP
+\fB\-\-sortStrategy\fR
+Read sortiing strategy
+["longest_and_strand_balanced"]
+.TP
+\fB\-\-minPoaCoverage\fR
+Minimum number of reads required
+within a window to call consensus and
+variants using arrow or poa. [3]
+.SH OPTIONS
+.TP
+\fB\-h\fR,\-\-help
+Output this help.
+.TP
+\fB\-\-log\-level\fR,\-\-logLevel
+Set log level. ["INFO"]
+.TP
+\fB\-\-version\fR
+Output version info.
+.TP
+\fB\-\-emit\-tool\-contract\fR
+Emit tool contract.
+.TP
+\fB\-\-resolved\-tool\-contract\fR
+Use args from resolved tool
+contract.
+.SS "Arguments:"
+.TP
+INPUT
+The input BAM alignment file
+.SH AUTHOR
+This manpage was written by Andreas Tille for the Debian distribution and can be used for any other usage of the program.
=====================================
debian/manpages
=====================================
@@ -0,0 +1 @@
+debian/*.1
=====================================
debian/patches/do_not_refer_to_non-existing_images.patch
=====================================
@@ -0,0 +1,28 @@
+Author: Andreas Tille <tille at debian.org>
+Last-Update: Wed, 10 Oct 2018 09:15:24 +0200
+Description: When trying to solve the potential privacy breach issue and
+ downloading these images I noticed that these do not exist any more.
+ Since I did not found any replacement I just removed those references
+
+--- a/doc/PBCCS.md
++++ b/doc/PBCCS.md
+@@ -2,10 +2,6 @@
+ pbccs - Generate Accurate Consensus Sequences from a Single SMRTbell
+ </h1>
+
+-<p align="center">
+- <img src="http://www.evolvedmicrobe.com/CCS.png" alt="Image of SMRTbell"/>
+-</p>
+-
+ ### Input
+
+ The ccs program needs a .subreads.bam file containing the subreads for each SMRTbell sequenced. Older versions of the PacBio RS software outputted data in bas.h5 files, while the new software outputs BAM files. If you have a bas.h5 file from the older software, you will need to convert it into a BAM. This can be done with the tool bax2bam which simply needs the name of any bas.h5 files to convert and the prefix of the output file. Assuming your original file is named mydata.bas.h5, you can produce a file mynewbam.subreads.bam with the following command.
+@@ -135,8 +131,6 @@ The Z-score for a subread is a metric wh
+
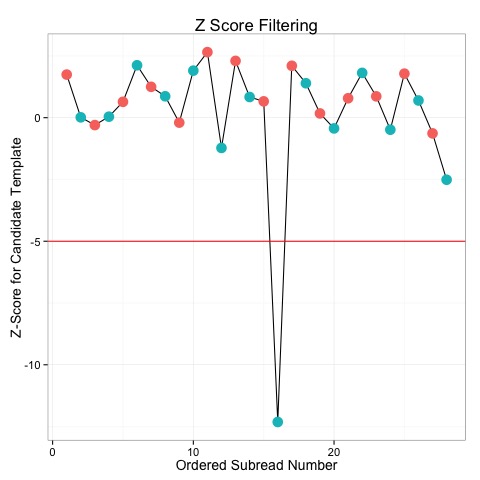
+ Subreads with very low Z-scores are very unlikely to have been produced according to the CCS model, and so represent outliers. For example, the plot below shows the Z-scores for several subreads. With a -5 cutoff, we can see that one subread is excluded from the data.
+
+-
+-
+
+ ## CCS Yield Report
+
=====================================
debian/patches/series
=====================================
@@ -1,3 +1,4 @@
add_missing_library_files
git-version.patch
no-static-linking.patch
+do_not_refer_to_non-existing_images.patch
=====================================
debian/rules
=====================================
@@ -16,6 +16,8 @@ export PYBUILD_NAME=consensuscore2
# Don't let pybuild consider using cmake
export PYBUILD_SYSTEM=distutils
+export DEB_BUILD_MAINT_OPTIONS=hardening=+all
+
docs = $(addprefix doc/,\
JULIET.html \
PBCCS.html \
View it on GitLab: https://salsa.debian.org/med-team/unanimity/compare/e00a110340e6cae7f249669daee69b5e3e5d521d...2eaa1f1af76e2758c0dfe36f3d578293dd6f72a4
--
View it on GitLab: https://salsa.debian.org/med-team/unanimity/compare/e00a110340e6cae7f249669daee69b5e3e5d521d...2eaa1f1af76e2758c0dfe36f3d578293dd6f72a4
You're receiving this email because of your account on salsa.debian.org.
-------------- next part --------------
An HTML attachment was scrubbed...
URL: <http://alioth-lists.debian.net/pipermail/debian-med-commit/attachments/20181011/58c22a8b/attachment-0001.html>
More information about the debian-med-commit
mailing list